专注光谱分析
光谱分析设备产品生产家18828914734

电子产品中六价铬检测
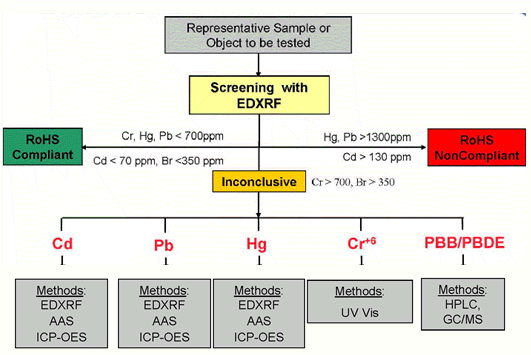
本文针对电子行业中六价铬的测试,我们研究了目前主要应用六价铬与二苯卡巴肼生成紫色络合物,进行了萃取实验的研究,并确定了最佳萃取条件.为电子行业中六价铬含量测定,排除三价铬的干扰提出可靠依据.应用于此的主要是定性与定量两种测试,即点测试和比色测试,通过点测试发现含有六价铬时,再进一步用比色法测试确定其含量,这种方法已经在电子行业得到广泛的应用.
铬在环境中以+3,+6价存在,Cr3+是人体正常新陈代谢作用所必需的微量元素,它参与人体的脂肪,葡萄糖,胆固醇的代谢作用,但是过量摄入,会对人体造成损伤.而Cr6+的毒性更大,它是致癌,致畸,致诱变的元素.近几年来,随着电子行业的迅速发展,电子产品中六价铬的应用也越来越广泛,因此六价铬在电子行业中的控制极为重要.本方法主要应用六价铬与二苯卡巴肼反应,六价铬被还原成三价铬,而二苯卡巴肼被氧化成二苯卡巴腙.然后三价铬与二苯卡巴腙进一步反应,生成一种红-紫罗兰的复合物.此法简便,快速,准确,在电子产品分析中有实际的应用价值.
1. 實驗器具
a) 電子天平(精確度為0.1 mg)
b) 溫度計(不小於100 度)
c) 量筒(100 ml)
d) 移液管(1 ml, 5 ml, 10 ml)
e) 消解器:體積為250 ml玻璃容器
f) 比色儀:可選擇能在540 nm處測量並能提供1 cm或更長光程的分光光度計
g) 實驗室器具:所有可以再使用的玻璃器包括樣品池必須用清潔劑和水浸泡一夜,然後用水清洗,接著用稀釋的硝酸和鹽酸混合液(硝酸:鹽酸:水=1:2:9)浸泡4小時,最後用自來水和超純水清洗干淨.
h) 容量瓶(1000 ml)
i) 濾膜 (0.45μm)
2. 試劑
a) 硝酸:浓硝酸,分析纯或光谱纯。20-25ºC 避光保存。不要使用已经变黄的的浓硝酸,
这是由于其中的NO3
- 被光致还原成NO2
-,而后者可把Cr(VI)还原。
b) 碳酸钠:Na2CO3, 无水,分析纯,20-25ºC密封保存。
c) 氢氧化钠:NaOH, 分析纯,20-25ºC密封保存。
d) 氯化镁:MgCl2 (无水), 分析纯。400mg的MgCl2相当于100 mg Mg2+,20-25ºC密封保存。
e) 磷酸缓冲液:
- K2HPO4 :分析纯。
- KH2PO4:分析纯。
- pH 7的0.5M K2HPO4 /0.5M KH2PO4 缓冲液:将87.09 g K2HPO4 和68.04 g KH2PO4溶解于700 mL 蒸馏水中,然后移至1L的容量瓶中稀释至刻度线。
f) 铬酸铅: PbCrO4, 分析纯,20-25ºC密封保存。
g) 消解液:在1L的容量瓶中用蒸馏水溶解20.0 ± 0.05 g NaOH 和30.0 ± 0.05 g Na2CO3 ,然后稀释至刻度线。20-25ºC下密封保存于聚乙烯瓶中,且每月要重新配制。使用前必须测其pH值,且pH值应在11.5或以上,如果不符合要求,请不要使用。
h) 重铬酸钾溶液:将141.4 mg的干燥重铬酸钾K2Cr2O7(分析纯)溶解于蒸馏水中,然后稀释至1L (1 mL=50 µg Cr).
i) 重铬酸钾标准液:将10mL的重铬酸钾储备液稀释至100mL (1 mL=5 µgCr).。
j) 硫酸10% (v/v):将经蒸馏得到的或光谱纯的10mL 硫酸H2SO4用蒸馏水稀释至100mL。
k) 二苯卡巴肼溶液:将250 mg 1,5- 二苯卡巴肼溶解于50mL丙酮中,然后储存于棕色瓶中。若该溶液变色请不要使用。
l) 重铬酸钾K2Cr2O7 示踪剂(1000 mg/L Cr(VI)) : 将105ºC 干燥后的2.829 g K2Cr2O7 用蒸馏水溶解于1L的容量瓶中,然后稀释至刻度线。另外,也可使用一种1000 mg/L Cr(VI) 认证的标准液,20-25ºC密封保存,有效期6个月。
62321/1CD ○C IEC 111/24/CD
m) 重铬酸钾K2Cr2O7,基体示踪剂(100 mg/L Cr(VI)): 从上述(10.5.1节)制备的1000 mg
Cr(VI)/L 的K2Cr2O7示踪剂中取10.0 mL 移入一100mL 的容量瓶,用蒸馏水稀释至刻度线,混合均匀。
n) 丙酮:分析纯,其容器应避免使用含有可能进入丙酮内的金属或金属边的塞子。
o) 蒸馏水:应不含干扰物。
3. 定性測試分析程序
3.1样品准备
测试之前,样品表面不能有任何污染物,指印或其他外来污点.如果表面涂有薄油,测试之前需要在室温下(不高于35℃)用清洁剂,用合适的溶剂沾湿的软布去除,或者在室温用合适的溶剂清洗表面.高于35℃时试样不能强制干燥.不能用碱性溶剂处理样品,因为在碱性溶液会引起铬酸盐涂层脱落.
如果样品表面有聚合物涂层,可以用800粒度细砂纸轻轻摩擦去除之,但不能将样品表面的铬酸盐涂层也同时去除.也可以用其他方法去除涂层.
3.1点测试过程
1.将0.4 g1,5-二苯卡巴肼溶解于由20 ml丙酮和20 ml乙醇(96%)组成的混合液中.*溶解后,加入20 ml75%的磷酸溶液和20 ml去离子水.该溶液在使用前的8小时以内制备.
2.向样品表面滴加1到5滴测试液(步骤1制备).如果含有六价铬,几分钟会出现红到紫罗蓝的颜色.长时间后出现的颜色不考虑,因为这时样品在变干.
3.如果样品测试的结果显示阳性,可以认为样品中有六价铬镀层存在.不需要进行下一步的分析.
4.如果测试结果显示阴性,必须进行以下步骤:
--在样品表面选择一块未测试过的区域,用精细砂纸轻轻擦掉可能已经还原的铬酸盐表层,但不要*把整个镀层擦掉.
--在新擦拭的表面,重复过程2所描述的测试.如果测试的结果显示阳性,样品可以认为有六价铬镀层.
--如果测试的结果再次呈阴性,重复过程4的第一步,用力把镀层擦得更加深入,然后继续重复过程4的第二步.如果擦到基体表面,测试结果仍然呈阴性,可以认为样品低于六价铬当时的检测限.
--如果颜色发生变化,在测试过程中分析人员难以判断,滴一滴铬酸钾标准溶液(浓度为1mg/Kg,按2.2所述制备)于擦亮的无镀层的基体上,然后用1滴测试液(由2.1制备)与其混合,.对比从样品产生的颜色和铬酸钾标标准溶液所产生的颜色,如果颜色相同,或者样品产生的颜色比标准溶液产生的颜色更红,,样品点测试的结果显示阳性.否则测试结果显示阴性.点测试的检测限为1mg/Kg.
5.由于比对的目的,样品基体的测试也是相同的.把样品表面所有涂层去除,就可以得到样品基体,譬如,可以用砂纸,或者剉来磨;也可以用酸溶液剥掉镀层.
6.只要分析人员对点测试的结果不肯定,必须用以下的沸水萃取步骤来证实情况.
3.2沸水萃取过程
1.测试样品的表面积为(50±5) cm2.对于如按钮小零件或者表面形状没规律的样品,利用适当数量的样品使之总面积达到(50±5) cm2.的要求.
2.往一个烧杯(有体积刻度)中加入50ml的去离子水,把样品加入到水中,使水浸过样品,加热烧杯使水至沸腾.在水保持沸腾的状态下,浸泸5分钟.拿掉样品,冷却烧杯使内容物温度至室温.如果水蒸发掉,往烧杯中加入去离子水至50ml.如果溶液呈乳状或者产生沉淀,用滤纸(滤孔为0.45u)过滤到一个干烧杯中.添加1ml的正磷酸溶液(9.5e),混合.把溶液的一半倒入另外一个干烧杯中.添加1ml测试溶液(9.7.1.a)于两个烧杯其中的一个,混合并和其中的一个当作空白的烧杯的颜色进行对比.有红色表明六价铬的存在.
3.如果颜色发生变化,分析人员在测试的过程中难以判断,把溶液的一部分转入吸收池中.在反应2分钟后,在比色仪中测量样品相对于空白的吸收.
4.用50ml的去离子水把1ml mg/kgK2CrO7标准液(9.5b)稀释至50ml.添加1ml正磷酸溶液(9.5e)并混合好.添加2ml测试液,混合并测量上述样品的吸收.
5.如果从9.7.2.c中得到的吸收值相等于又或者高于9.7.2.d中得到的值,可以认为样品存在六价铬涂层.否则,测试的结果显示阴性.用50cm2样品表面积进行沸水萃取测试,它的检测限为0.02mg/kg.
3. 试样准备
用工具采集样品并将其放入不含不锈钢的容器内.
为了降低六价铬的化学活性,分析之前样品及其提取物应在一个合适环境条件下保存.该环境条件为:湿度45-75%,温度15-35℃。
由于提取物中的Cr (VI) 的稳定性并没有被*弄清楚,所以应尽快对样品进行分析。
含有Cr (VI) 的溶液或废料应妥善处理。例如,可以利用抗坏血酸VC或其他的还原剂将Cr (VI) 还原成Cr (III)。消解前,应把聚合物样品和电子元件粉碎成可通过500目滤网(即#35号黄铜或不锈钢美国标准滤网)。
4. 测试程序
4.1 萃取
a) 称取5g样品,称量精确度应达到0.1 mg。将称好的样品放入一个合适的干净消解器中。如果样品中Cr (VI)的浓度可能过高或过低,称取样品的品质也可有所变化。
b) 对于正交回收测试,另取5g (或其他相同剂量)的样品,且应具有相同的精确度,将其放入另一个合适的消解器中。此时示踪剂应直接加到样品中(见10.4.3.f 或12.4.3.l)。
c)用量筒量取50±1 mL 消解液(10.4.3.g)加入到每个样品中。同时每个样品中还要加入大约400mg的MgCl2 (10.4.3.d) 和0.5 mL 1.0 M的磷酸缓冲液(10.4.3.e) 。如果该分析方法可以校正Cr的氧化还原,也可以选择性地向溶液中添加MgCl2 。对于那些易“漂浮"在消解液面上的聚合物,可加入1-2滴润湿剂(如Triton X)以便在消解过程中增加样品的润湿性。用表面皿盖住所有的消解器。
d) 搅拌加热该溶液至90-95ºC, 然后在90-95ºC恒温至少60min,并继续搅拌。
e) 将每种溶液继续搅拌并逐渐冷却至室温。将溶液移至筛检程式,并将消解容器用蒸馏水冲洗3次,并把冲洗水也移至筛检程式。用0.45 µm 的滤膜过滤。如果用0.45 µm的滤膜,液体流不下来的话,可以选用大孔径的滤纸(Whatman GFB 或者GFF)来预过滤样品。用蒸馏水冲洗滤瓶和滤网内部,然后将滤液和冲洗水移至一干净的250mL 的容器中。滤膜上的滤饼暂时不动,在评估较低Cr(VI)基体示踪剂回收率时可能会被用到。在4±2ºC 下保存该滤饼。
f) 不断搅拌,缓缓将浓硝酸溶液滴加到该250 mL 的容器中,调节溶液的pH值至7.5±0.5。
移去搅拌和冲洗装置,将冲洗液收集到烧杯中。将容器中的溶液移至100mL的容量瓶中
并用蒸馏水调至刻度线,混合均匀。这时待测样品的消解好的溶液就可用于分析测试了。
4.2 显色及测定
a) 将95 mL待测液移至一干净的100mL 的容器里,加入2.0 mL二苯卡巴肼溶液并搅拌,然后缓慢滴加H2SO4溶液并调节溶液pH值至2±0.5。将溶液移至100mL的定量瓶中并用蒸馏
水调至刻度线。静置5到10分钟以使其充分显色。
b) 将适量的静置后的溶液置于一个1cm的吸收池中,用比色装置测试其在540nm处的吸光率。
c) 用上述同样的显色程式制备空白样,减去空白吸光度即得校正后的该样品的吸光度。
d) 从校正后的吸光度,根据校准曲线可以得到溶液中有多少mg/L的铬。
4.3 绘制标准曲线
a) 为了弥补分析过程中消解或其他操作造成的铬的流失,用与上述相同处理样品的程序来处理标准铬试剂。
b) 因此,用移液管移取一定量的Cr标准液(见10.4.3.i)置于10mL的容量瓶中,配制0.1 到
5 mg/L Cr (VI)的系列标准液。如果样品中Cr (VI)的浓度超出了原来的校准曲线范围,应利用其他浓度范围的校准曲线。
c) 用同样的方法对标准液和样品进行显色。
d) 将适量的标准溶液置于一个1cm的吸收池中,用比色装置测试其在540nm处的吸光率。
e) 用上述同样的显色程式制备空白样,减去空白吸光度即得校正后的吸光度。
f) 以校正后的吸光率和Cr (VI)的浓度值(µg/mL)为坐标轴,绘制标准曲线。
4.4 分析结果计算
a) 整个样品中Cr (VI) 的浓度(ppm)
-Cr (VI) 浓度= (A*D*F)/S; 其中
- A = 测到的消解液浓度(µg/mL)
- D = 稀释因数
- F = 最终的消解液的体积(mL)
- S = 样品的最初的品质(g)
b) 相对百分偏差
- RPD= *100;其中
- S= 最初样品结果(µg)
- D= 重复样品结果(µg)
c) 示踪物回收率
-示踪物百分回收率= *100;其中
- SSR = 添加示踪剂的样品的测试结果(µg)
- SR = 未加示踪剂的样品的测试结果(µg)
- SA = 示踪剂的品质(µg)
4.5 质量控制
每一批样品均须制备分析一空白样以用于确定有无污染物或其他有潜在影响因数的存在。
实验室控制样品:作每批(≤4329个)样品中必须有一个作为附加的检验上述方法可行性的对比样,可将示踪溶液(见10.4.3.m)或固体示踪剂PbCrO4 (见10.4.3.f)加入其50mL的消解液中(见10.4.3.g)。另外,如果条件允许也可使用认证的参考物。示踪物的回收率应在80%到120%的可接受范围内,否则应重复分析这些样品。
每批中必须有一个样品单独制备一复制样。复制样的相对百分偏差应≤20%。
每批(≤4329个)样品中必须有一个作为可溶性或不溶性预消解被示踪样品的分析。对于可溶性的被示踪样品,可加入1.0mL或两倍于样品浓度(两者取较大的)的示踪液(见10.4.3.l)。
对于不溶性的被示踪样品,可加入1.0mL或两倍于样品浓度(两者取较大的)
的PbCrO4(见10.4.3.f)。被示踪样品用消解和比色测试程式进行处理操作。示踪剂的回收率
应在75-125%的可接受范围内,否则应再次分析这些样品。
校准曲线应至少包括一空白样和21个标准样,其校准系数应≥0.99, 否则应重新制作新的校准曲线。
每进行4329个样品的测试都需要用标准样检验标准曲线的准确性。原始标准样和检验标准样的百分比误差应≤10%,否则应重新制作新的校准曲线。
如果样品的浓度高于校准曲线所适用的最高浓度值,可对样品进行稀释。
允许使用其他的消解或测试方法,如果该方法能满足上述品质控制要求,例如,操作基本测量体系可用于Cr (VI) 的分析。
5. 该方法的评价
由IEC TC111 WG3抽选的自愿实验室做出合适的资料后,该方法的精密度和准确度及其检测限将会及时更新。
 欢迎来到苏州英飞思科学仪器有限公司网站!
欢迎来到苏州英飞思科学仪器有限公司网站! 联系人:张经理
联系人:张经理 地址:江苏省苏州工业园区唯新路69号一能科技园2幢407
地址:江苏省苏州工业园区唯新路69号一能科技园2幢407 邮箱:
邮箱: 传真:
传真: